深势科技 DP Technology:Uni-FEP上线公测——旧时王谢堂前燕,飞入寻常百姓家Hermite,是深势科技推出的根植于云计算的临床前计算机辅助药物设计(Computer Aided Drug Design, CADD)平台,致力于为药物研发工作者提供在CADD中数据、算法、算力三位一体的一站式解决方案。 Uni-FEP是Hermite最新推出的药物结合自由能计算模块,将自由能微扰理论、分子动力学、增强采样算法与高性能计算相结合,能够以化学精度高效评估蛋白质与配体的结合亲和力,实现工业规模的先导化合物优化。 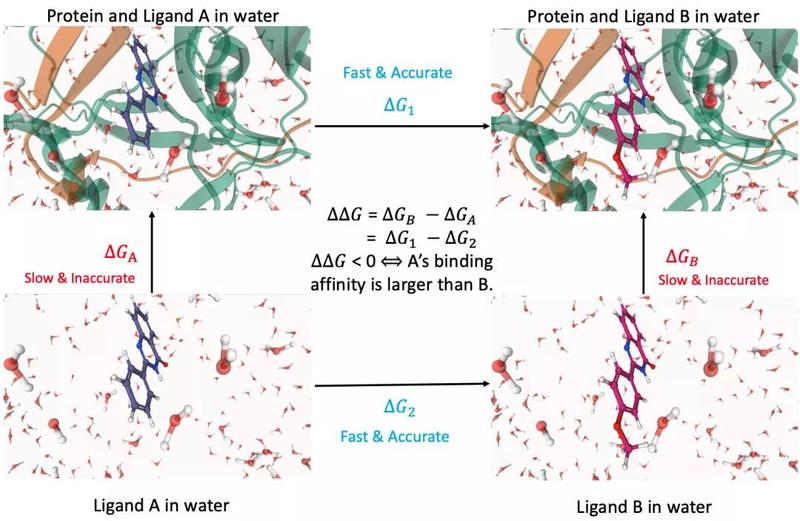 性能强大,高效准确 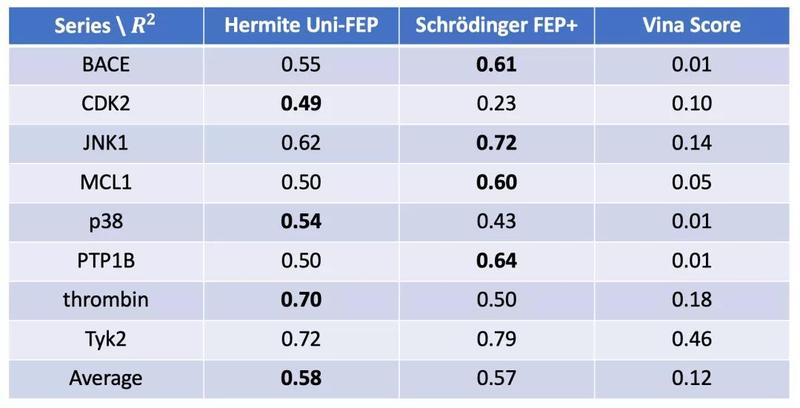 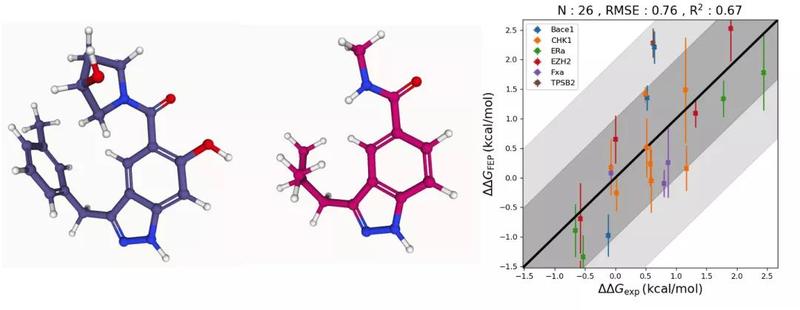 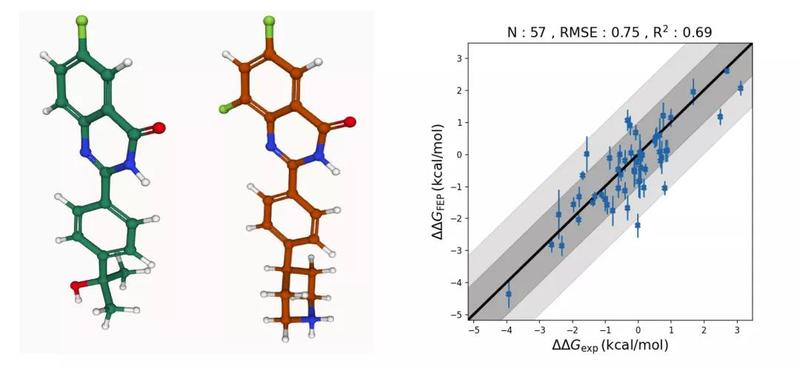 流程自动,操作便捷
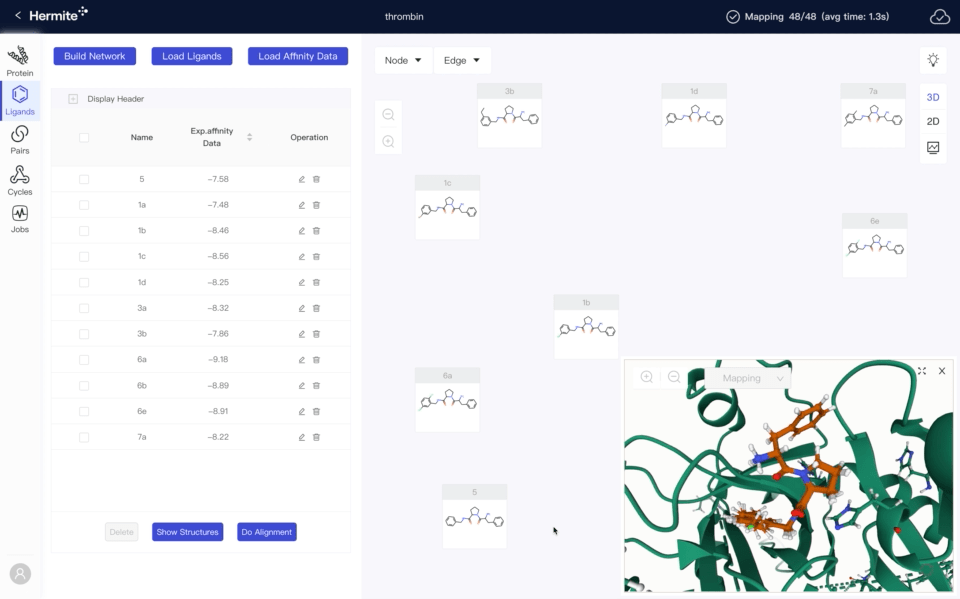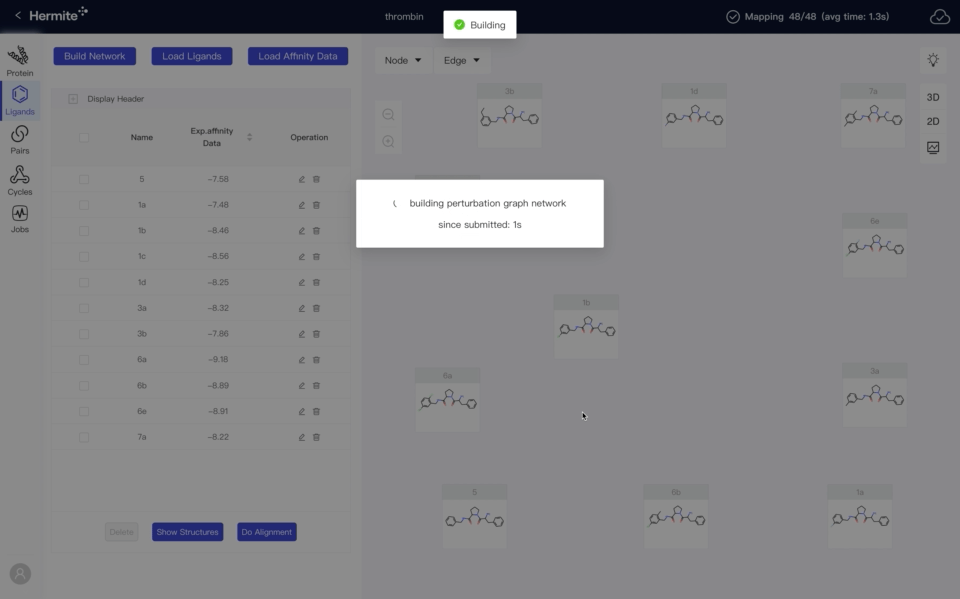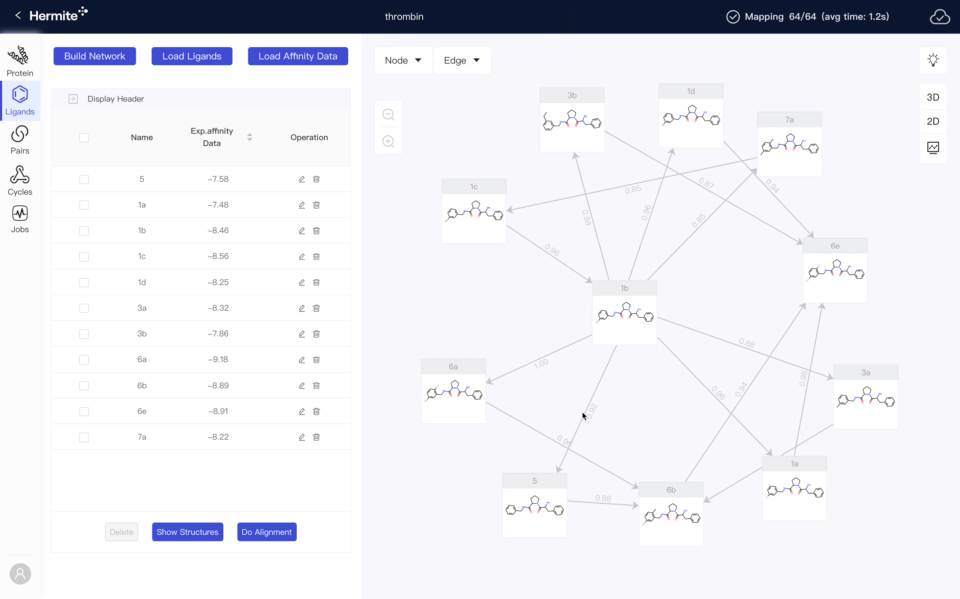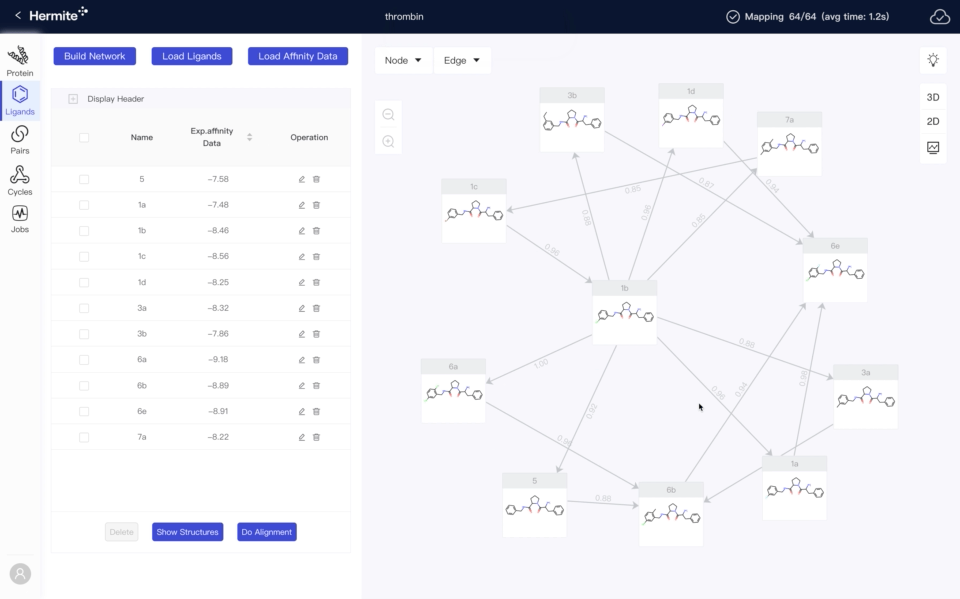 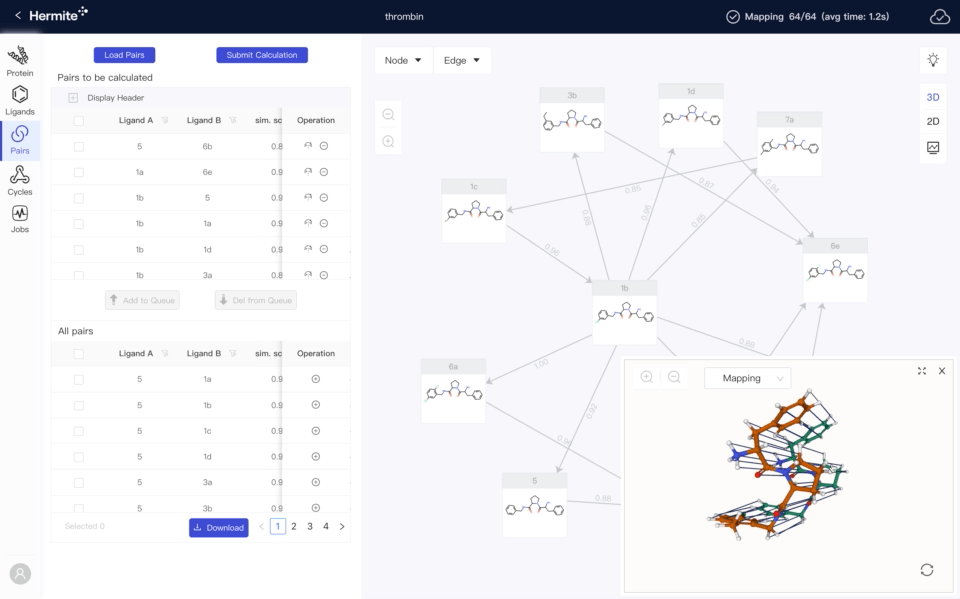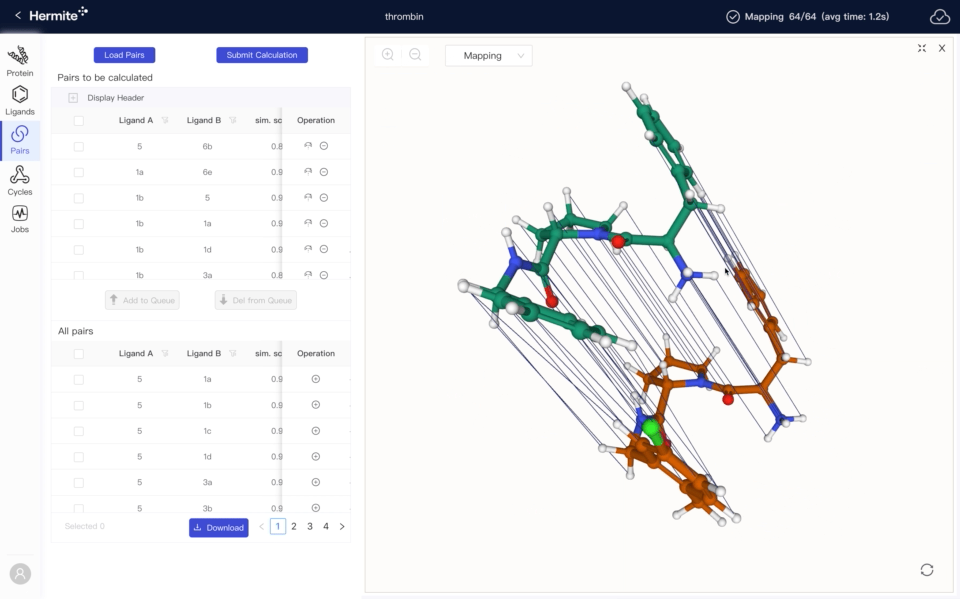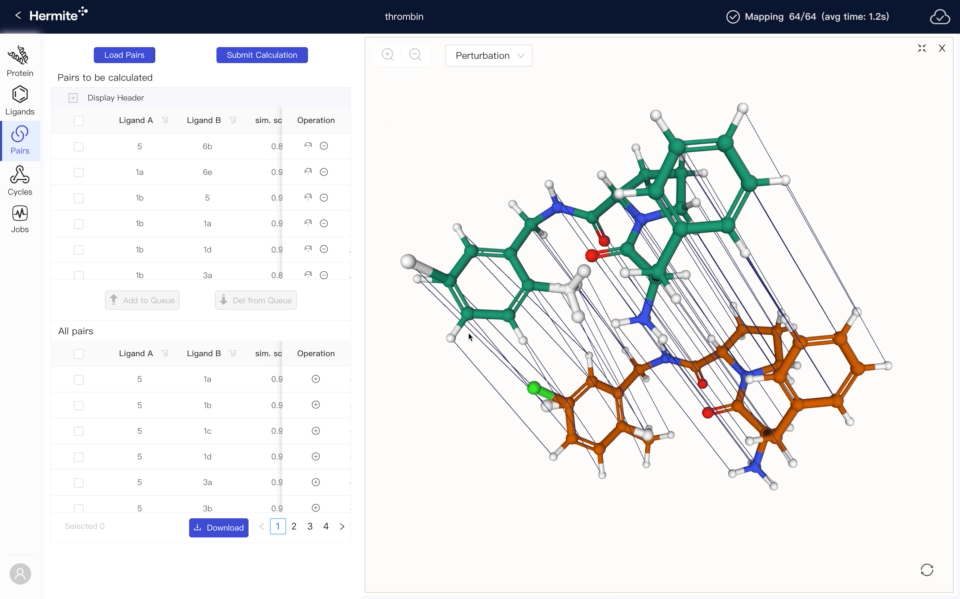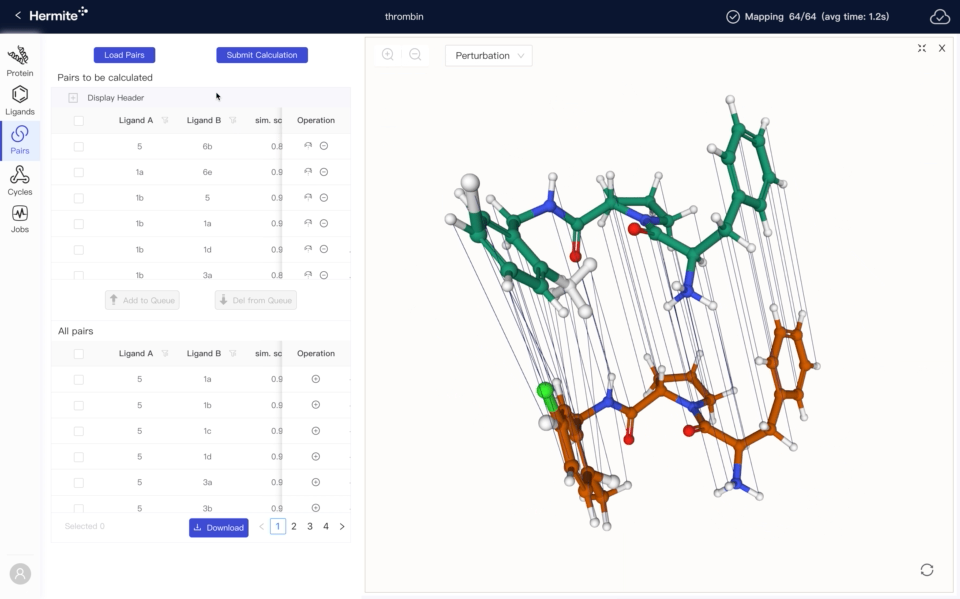 需求导向,快速迭代
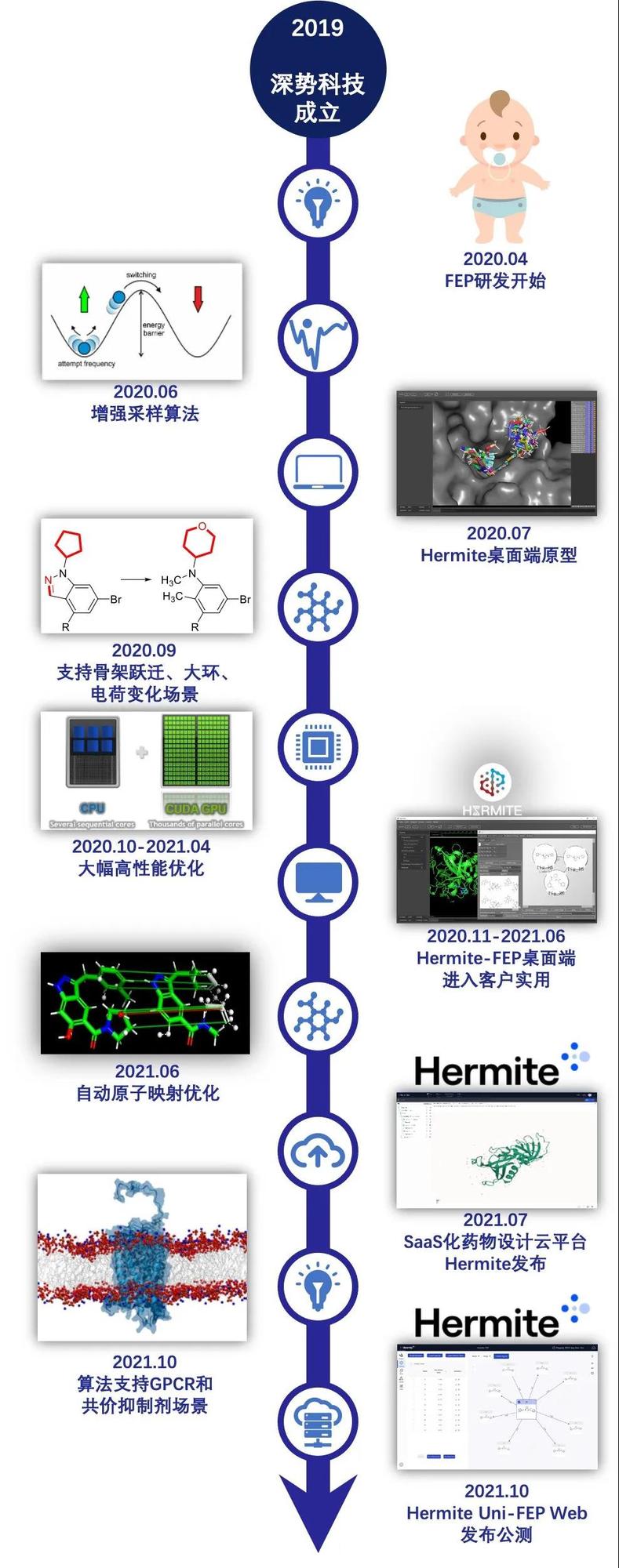
图7: Uni-FEP研发历程  References [1] Lengauer, Thomas, and Matthias Rarey. "Computational methods for biomolecular docking." Current opinion in structural biology 6.3 (1996): 402-406. [2] Chipot, Christophe, and Andrew Pohorille. "Free energy calculations." Springer series in chemical physics 86 (2007): 159-184. [3] Jorgensen, William L. "Efficient drug lead discovery and optimization." Accounts of chemical research 42.6 (2009): 724-733. [4] Wang, Lingle, B. J. Berne, and Richard A. Friesner. "On achieving high accuracy and reliability in the calculation of relative protein–ligand binding affinities." Proceedings of the National Academy of Sciences 109.6 (2012): 1937-1942. [5] Cournia, Zoe, Bryce Allen, and Woody Sherman. "Relative binding free energy calculations in drug discovery: recent advances and practical considerations." Journal of chemical information and modeling 57.12 (2017): 2911-2937. [6] Wang, Lingle, et al. "Accurate and reliable prediction of relative ligand binding potency in prospective drug discovery by way of a modern free-energy calculation protocol and force field." Journal of the American Chemical Society 137.7 (2015): 2695-2703. [7] Bennett, Charles H. "Efficient estimation of free energy differences from Monte Carlo data." Journal of Computational Physics 22.2 (1976): 245-268. [8] Van Der Spoel, David, et al. "GROMACS: fast, flexible, and free." Journal of computational chemistry 26.16 (2005): 1701-1718. [9] Wang, Lingle, et al. "Modeling local structural rearrangements using FEP/REST: application to relative binding affinity predictions of CDK2 inhibitors." Journal of chemical theory and computation 9.2 (2013): 1282-1293. [10] Bussi, Giovanni. "Hamiltonian replica exchange in GROMACS: a flexible implementation." Molecular Physics 112.3-4 (2014): 379-384. [11] Wang, Lingle, et al. "Accurate modeling of scaffold hopping transformations in drug discovery." Journal of chemical theory and computation 13.1 (2017): 42-54. [12] Schindler, Christina EM, et al. "Large-scale assessment of binding free energy calculations in active drug discovery projects." Journal of Chemical Information and Modeling 60.11 (2020): 5457-5474. [13] Zhang, Linfeng, et al. "Deep potential molecular dynamics: a scalable model with the accuracy of quantum mechanics." Physical review letters 120.14 (2018): 143001. [14] Zhang, Linfeng, et al. "End-to-end symmetry preserving inter-atomic potential energy model for finite and extended systems." Advances in Neural Information Processing Systems (2018): 4441-4451. [15] Zhang, Yuzhi, et al. "DP-GEN: A concurrent learning platform for the generation of reliable deep learning based potential energy models." Computer Physics Communications 253 (2020): 107206.
文章分类:
对外活动
|